MS Reactivity
Automated workflows for design, optimization, and unsupervised mechanism discovery in molecular chemistry

Automated workflows for design, optimization, and unsupervised mechanism discovery in molecular chemistry
MS Reactivity offers a comprehensive suite of computational capabilities that enable highly automated workflows for molecular (catalyst) design, reaction optimization, and unsupervised mechanism discovery in molecular chemistry. Its two flagship tools are Reaction Network Enumeration Profiler (RxnEnumProfiler) and Nanoreactor. Complementing these is CREST GUI, a user-friendly interface for CREST – a utility and driver program for the semiempirical quantum chemistry package xTB.
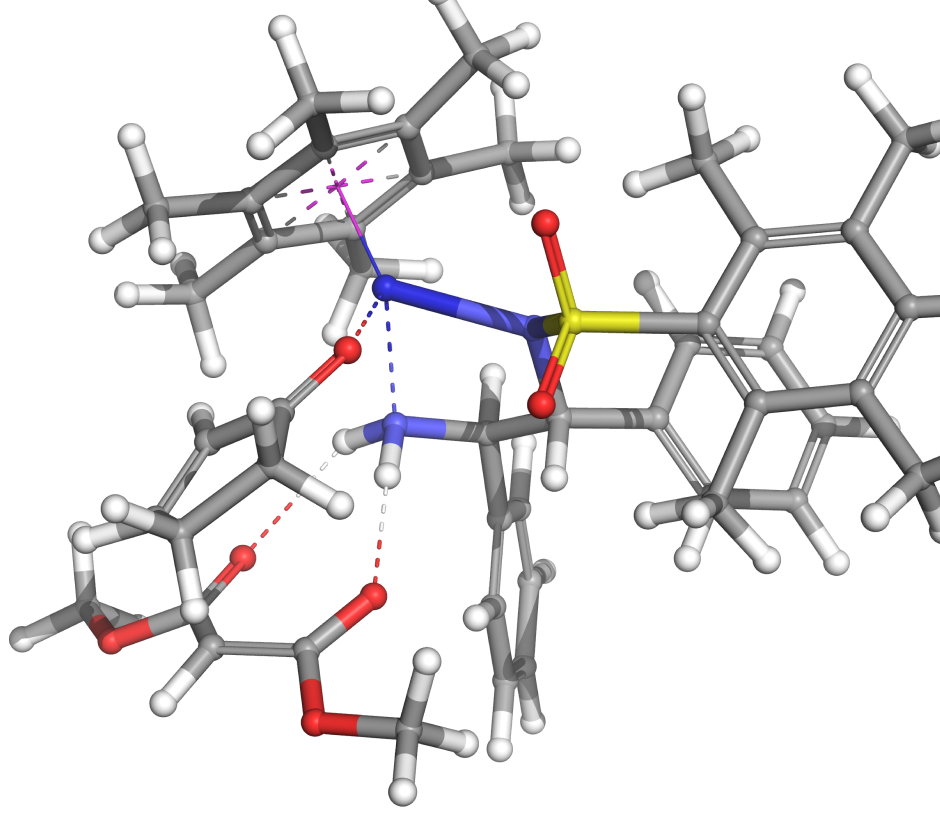
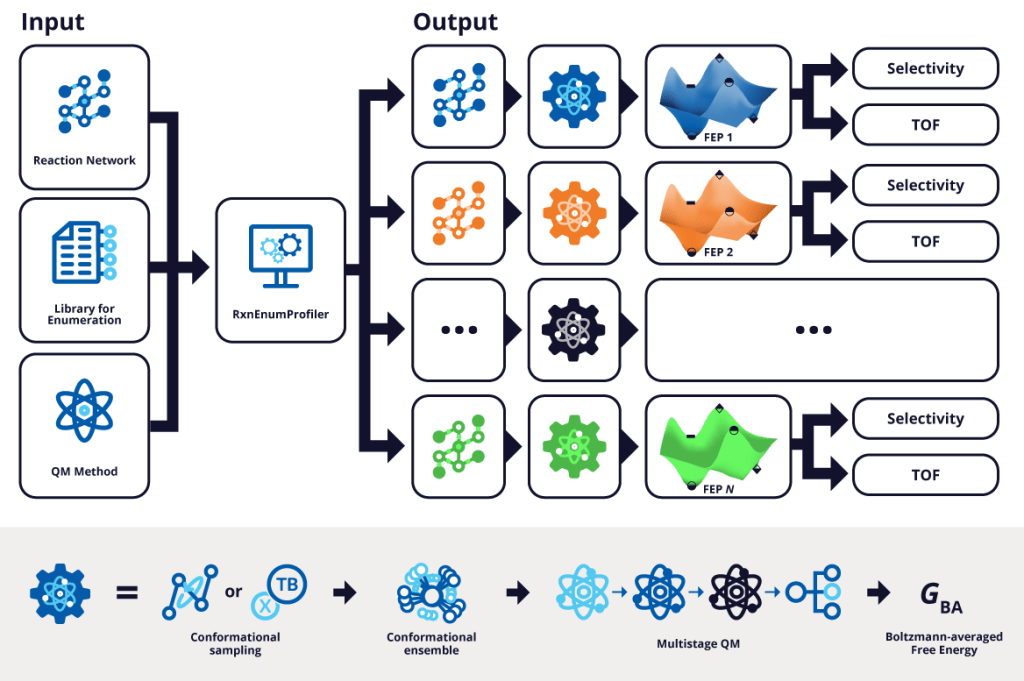
Virtual high-throughput screening (vHTS) of reaction networks is a computational strategy for systematically evaluating large libraries of chemical species within a fixed reaction topology—that is, a predefined sequence of mechanistic steps involving reactants, products, intermediates, and/or transition states that characterize a catalytic or chemical process. RxnEnumProfiler is a fully automated, massively parallel workflow specifically developed to enable this process. It functions by automatically enumerating a user-defined reference reaction network and computing the corresponding free energy profiles (FEPs). These profiles are calculated as either Boltzmann-averaged conformational ensemble Gibbs free energies (GBA) or lowest-energy conformer G values, based on a specified quantum mechanical level of theory.
Automated reaction discovery lies at the heart of predictive chemistry, enabling chemists to design chemical processes that are smarter, faster, cleaner, and more efficient. Nanoreactor – Elementary Reaction Network, a tool by Schrödinger, automates the identification of relevant elementary reactions starting from a known local minimum on the xTB potential energy surface (PES). Complementing this, Potential Energy Surface Sampling-Sorting enhances Nanoreactor’s capabilities by systematically exploring and ranking minima states on xTB (or DFT) PES. Since chemical reactions tend to follow the downhill path on the free energy surface, this feature focuses on pinpointing the most probable final products.


CREST GUI panel offers access to various algorithms implemented in CREST 3.0, a utility and driver program for the semiempirical quantum chemistry package xTB.1,2 The program’s name originated as an abbreviation for Conformer – Rotamer Ensemble Sampling Tool as it was developed as a program for conformational sampling at the extended tight-binding level GFN-xTB. Since then several functionalities have been added to the code. In its current state, the program provides a variety of sampling procedures, for example for improved thermochemistry, or explicit solvation.
References:
Discover how Schrödinger technology is being used to solve real-world research challenges.
Get answers to common questions and learn best practices for using Schrödinger’s software.

Learn more about the related computational technologies available to progress your research projects.
Quantum mechanics solution for rapid and accurate prediction of molecular structures and properties
A modern, comprehensive force field for accurate molecular simulations
Automated machine learning tools for materials science applications
Get more from your ideas by harnessing the power of large-scale chemical exploration and accurate
in silico molecular prediction.
Level up your skill set with hands-on, online molecular modeling courses. These self-paced courses cover a range of scientific topics and include access to Schrödinger software and support.
Learn how to deploy the technology and best practices of Schrödinger software for your project success. Find training resources, tutorials, quick start guides, videos, and more.